Gastrointestinal Cancers in a Peutz-Jeghers Syndrome Family: A Case Report
Article information
Abstract
A 17-year-old man was diagnosed as Peutz-Jeghers syndrome (PJS) because of pigmented lip and multiple gastrointestinal polyps. He had anemia and underwent polypectomy on the duodenum and colon. His maternal family members were patients with PJS. His mother used to be screened with endoscopy to remove large polyps. One and half years later, he underwent jejunal segmental resection due to intussusceptions. He underwent endoscopic polypectomy every 2 to 3 years. When he was 23 years old, high-grade dysplasia was found in colonic polyp and his mother underwent partial pancreatectomy due to intraductal papillary mucinous carcinoma. When he was 27 years old, diffuse gastric polyps on the greater curvature of corpus expanded and grew. Therefore, wide endoscopic polypectomy was done. Histological examination revealed focal intramucosal carcinoma and low-grade dysplasia in hamartomatous polyps. We report cases of cancers occurred in first-degree relatives with PJS.
INTRODUCTION
Hyperpigmented mucocutaneous macules are clues to the suspicion of Peutz-Jeghers syndrome (PJS). PJS is an inherited multiple gastrointestinal polyposis associated with mucocutaneous pigmentation. In 1921, Peutz1 first reported it, describing a Dutch family with gastrointestinal polyposis with pigmentations. Jeghers et al.2 also reported 10 cases from different families in 1949. It is caused by a germline mutation in the serine/threonine-protein kinase 11 (STK11) and inherited in an autosomal dominant manner.3
Prevalence of PJS is approximately one in 200,000. Polyps occur anywhere in the gastrointestinal tract and are characteristic hamartomas. They can cause abdominal pain, anemia and gastrointestinal bleeding by intussusception or obstruction. Although the mechanism of carcinogenesis in PJS is unknown, breast and gastrointestinal malignancies are often accompanied. Relative risk of pancreatic cancer in PJS was 132-folds higher than in general population.4 The number of affected first-degree relatives is associated with increased cancer risk.
We report here cases of cancer in first-degree relatives with PJS who had intraductal papillary mucinous carcinoma of pancreas in mother and intramucosal adenocarcinoma of stomach in son.
CASE REPORT
A 17-year-old male patient was diagnosed as PJS because of dark pigmented macules on the lips and multiple hamartomatous polyps in the stomach, duodenum, and colon. He had anemia and underwent polypectomy in the duodenum and colon. His maternal grandfather, mother, two uncles and two cousins were patients with PJS (Fig. 1). One uncle had died of lung cancer at the age of 32. His mother used to be screened with endoscopy to remove large polyps.
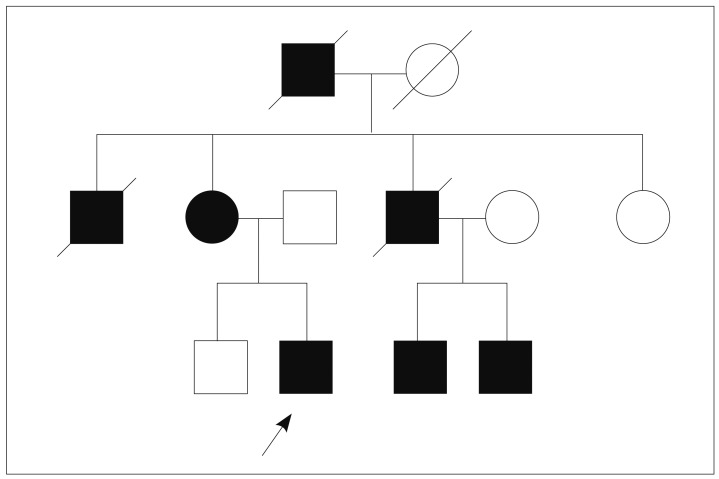
The pedigree of the patient. The arrow indicates the patient. His two uncles died of renal failure and lung cancer.
One and half years later, he underwent jejunal segmental resection due to intussusceptions. Thereafter, he underwent gastroduodenoscopy and colonoscopy every 2 to 3 years.
When he was 23 years old, colonoscopic polypectomy was done and high-grade dysplasia was found in colonic polyp. Simultaneously, his mother presented abdominal discomfort at the age of 51. Computed tomography (CT) scan showed a 3-cm sized multiseptated cystic lesion with enhanced nodular portion in the pancreas (Fig. 2). To make sure ductal communications of the lesion and to reveal the drainage of mucin, endoscopic retrograde cholangiopancreatography (ERCP) was done. ERCP showed a focal filling defect and extraductal collection of dye at the pancreatic body. Ultrasound-guided biopsy revealed intraductal papillary mucinous adenoma with low grade dysplasia and CA 19-9 in the aspirated fluid from the pancreas was elevated to 379.58 U/mL. Partial pancreatectomy was performed and intraductal papillary mucinous carcinoma in situ was confirmed (Fig. 3).
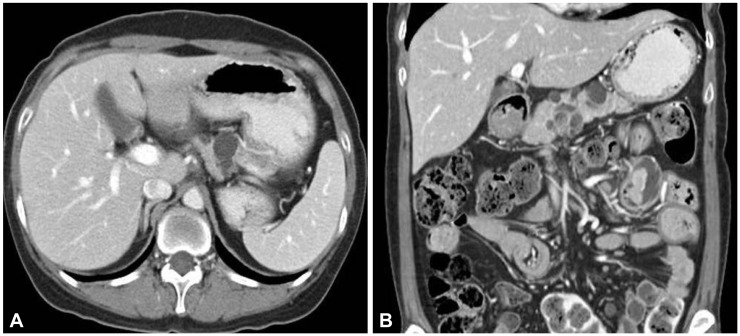
(A) Abdominal computed tomography scan. (B) Multiple variable sized and multiseptated cystic lesions are shown in the pancreas. Pedunculated polyps are found in the jejunum.
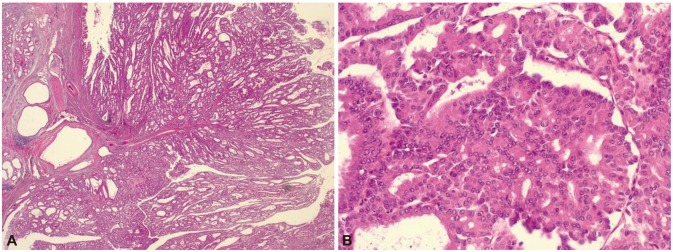
Histopathologic findings of the resected pancreas from the patient's mother. The intraductal papillary mucinous neoplasm with irregular branching papillae and cribriform growth showing architectural and cytological atypia (A, H&E stain, ×12; B, H&E stain, ×200).
When he was 27 years old, he suffered from abdominal discomfort and dizziness. His conjunctiva was pale and hemoglobin was 7.3 g/dL but other biochemical studies were normal. Gastroduodenoscopy revealed multiple papillary polyps diffusely covering the greater curvature of the corpus (Fig. 4). Wide endoscopic polypectomy was performed under general anesthesia. Histological examination revealed hamartomatous polyps with low-grade dysplasia and focal intramucosal carcinoma (Fig. 5). CT scan showed remnant massive polyps in the stomach. The patient was referred to surgical department for operation but subtotal gastrectomy was performed in another hospital following the opinions of his family.

(A) Endoscopic findings of the patient. (B) Diffuse nodular and papillary polyps covering the greater curvature of the body. Focal hyperemic large nodular polyps are shown.
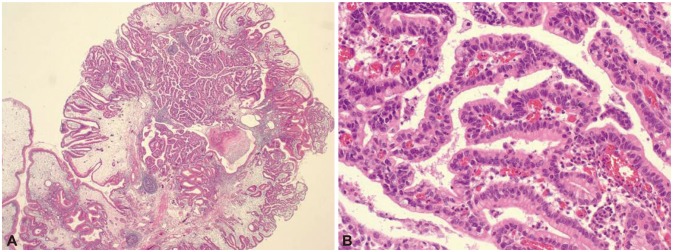
Histopathologic findings of the gastric polypectomy specimen. Gastric polyp shows villous morphology and branching multi-layered nucleus foci of carcinomatous change (A, H&E stain, ×12; B, H&E stain, ×200).
DISCUSSION
Polyps in PJS had typical hamartomatous polyps with arborizing pattern of smooth muscles proliferation.5 Although the mechanism of carcinogenesis and whether hamartomas are premalignant are unknown, PJS increased risks for the development of both gastrointestinal and extragastrointestinal malignancies. A hamartoma-adenoma-carcinoma pathway was suggested by the presence of adenomatous foci within polyps of PJS. However, several factors contradict this hypothesis. A total of 2,461 polypectomy was performed in 63 patients with a median follow-up of 10 years, and only six polyps contained atypia or dysplasia.6 Hamartomatous polyps were more common in the small intestine, but most gastrointestinal cancers were colorectal cancers.7
Germline mutations in STK11 are localized on chromosome 19p13.3 or 19q13.4 in PJS. STK11 regulates cellular proliferation via G1 cell-cycle arrest and has a role in cell polarity. Cancer in PJS develops through mucosal instability, which is associated with STK11 mutation. A pathogenic STK11 mutation was detected in 80% to 94% of the families with PJS.8
Since PJS is a rare disorder, it is difficult to assess risks of cancer in PJS. However, a few collected data were published. One of the studies reported that 23% of PJS patients developed cancers. The risk of cancer stratified by age increases rapidly after the age 50 for all cancers.9 Relative risk for all cancers was 15.2 in PJS. Relative risks and absolute rates for cancer in small bowel, stomach, pancreas, colon, esophagus, breast, and ovary were high.10
Surveillance in patients with PJS has two purposes. One is to detect early carcinomas and the other is to prevent morbidity such as bleeding, anemia, intussusceptions, and obstruction by removing large and eroded polyps. Endoscopic examinations are recommended every 3 years, beginning from the age of 18. Our patient underwent operation due to small bowel intussusception at the age of 19, even though he had had surveillance for polypectomy. Evaluation for breast starts from the age of 25 to 30 with annual ultrasound and mammography after the age of 50. Cost-effectiveness of screenings for pancreas and genital tract are still controversial.8
We reported a family with cancers that have developed in the first degree relatives with PJS. Despite the relative rarity of cancer incidence, surveillance for various cancers is an important part of management in patients with PJS.
Notes
The authors have no financial conflicts of interest.